
LEMMI
user guide
TIP
The platform is described in Seppey M, Manni M, Zdobnov EM. LEMMI: A continuous benchmarking platform for metagenomics classifiers [published online ahead of print, 2020 Jul 2]. Genome Res. 2020;gr.260398.119. doi:10.1101/gr.260398.119
https://genome.cshlp.org/content/early/2020/07/01/gr.260398.119 (opens new window)
# Table of content:
- Summary
- Terminology
- User interface
- Workflow
- Two kinds of reference
- Two category of benchmark
- Results assessment
- Scoring scheme
- Version control
- Tutorial: wrap your method into a container for benchmarking
- FAQ
# Summary
Metagenomics analyzes consist of multiple steps conducted first in the wet-lab and then on the e-lab. When designing a pipeline, one needs to select among all methods those that maximize the quality of the results given the resources available. Comparing entire workflows at once, from the sample preparation to the final computational predictions, shows how using different tools and methods lead to different results. However, to precisely identify the cause of the discrepancies, and which choices produce the best results, one needs to assess each brick of the pipeline separately, in an independent, reproducible, and controlled way. And to keep track of every new method in a fast evolving field, this has to be done continuously.
The LEMMI platform offers a container-based benchmark of methods dedicated to the step of taxonomic classification. This encompasses profilers and binners that rely on a reference to make their predictions. In LEMMI, a method configured with specific parameters and associated with a reference constitutes a configuration. The performances of configurations when analyzing several mock in-silico datasets are evaluated by comparing their predictions to the ground truth according to several metrics. Runtime and memory consumption are other factors measured as part of the performances of each configuration.
The Web interface of the LEMMI platform presents the evaluated configurations in different rankings. They can be customized by the user according to the importance (weight) they give to every metrics. Default presets are available for common experimental goals, and customized rankings are identified by a fingerprint that allows a user or method developers to share them so they can be reloaded through the url (opens new window) of the LEMMI platform.
To minimize the prediction differences resulting from the use of dissimilar references, which brings noise to the evaluation, the LEMMI platform exploits the ability of many methods to build a new reference using provided genomic content. It creates configurations for which the reference is various subsets of the RefSeq assembly repository. Additionally, when using datasets generated within the benchmarking platform, the genome exclusion approach is used, which simulates a situation closer to real life scenarios.
As users often choose standard software packages comprising a method bundled with a default reference, the LEMMI platform also evaluates such configurations, in which the reference is not built as part of the evaluation but provided with the method container.
# Terminology
# Container
Isolated software packages. See https://www.docker.com/resources/what-container (opens new window)
# Method
One tool, script, program or pipeline that functions as taxonomic classifier.
# Profiler
Taxonomic classifiers that produce relative abundance profiles, i.e. they quantify the proportion of each organism in the sample by taxonomic rank.
# Binners
Taxonomic classifiers that cluster reads into taxonomic bins, i.e. they assign a label (taxid) to each read.
# Parameter
Any value that can vary within a method algorithm. Can be the k-mer size, some sort of threshold, etc.
# References
The catalogs of known genomes that are used by the method to match the reads to known taxa.
# Configuration
The combination of a method, specific value for parameters, and a specific reference.
# BUNDLED
Stands for the kind of reference that are pre-packaged and submitted for evaluation with the method container.
# BUILT
Stands for the kind of reference that is created during the evaluation using genome files that are controlled by the LEMMI platform.
# Genome exclusion
BUILT+GE: An extension of the BUILT kind of reference, in which the source of the reads that are being evaluated is excluded from the reference that is going to be built for the analysis. It prevents an overfitted reference to be used and is used in the benchmark category "METHOD ALGORITHMS"
# Dataset
In silico generated sequencing reads, in fastq format.
# Prediction
The description of the data (binning and profiling) as reported by a configuration.
# Ground truth
The true description of the data.
# Score
A value that summarizes all weighted metrics describing the performance of the predictions over all datasets for a configuration.
# Fingerprint
A string identifier (e.g. SD.DEFAULT.beta01.20190417) assigned to each ranking that can be pasted in the URL to recover it at any time.
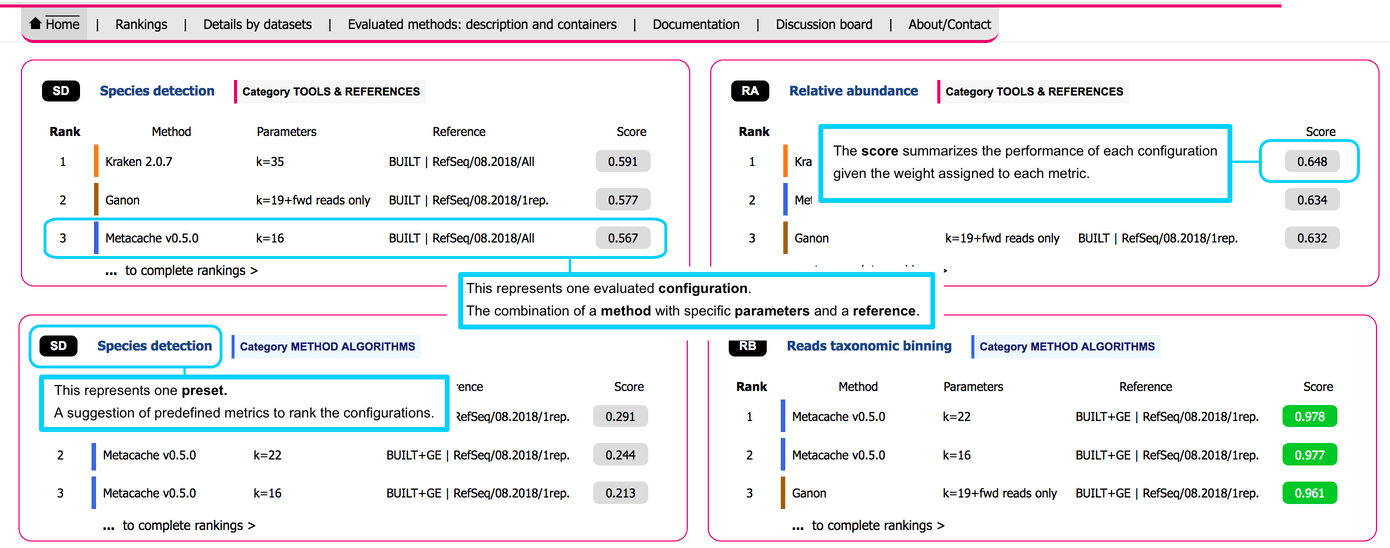
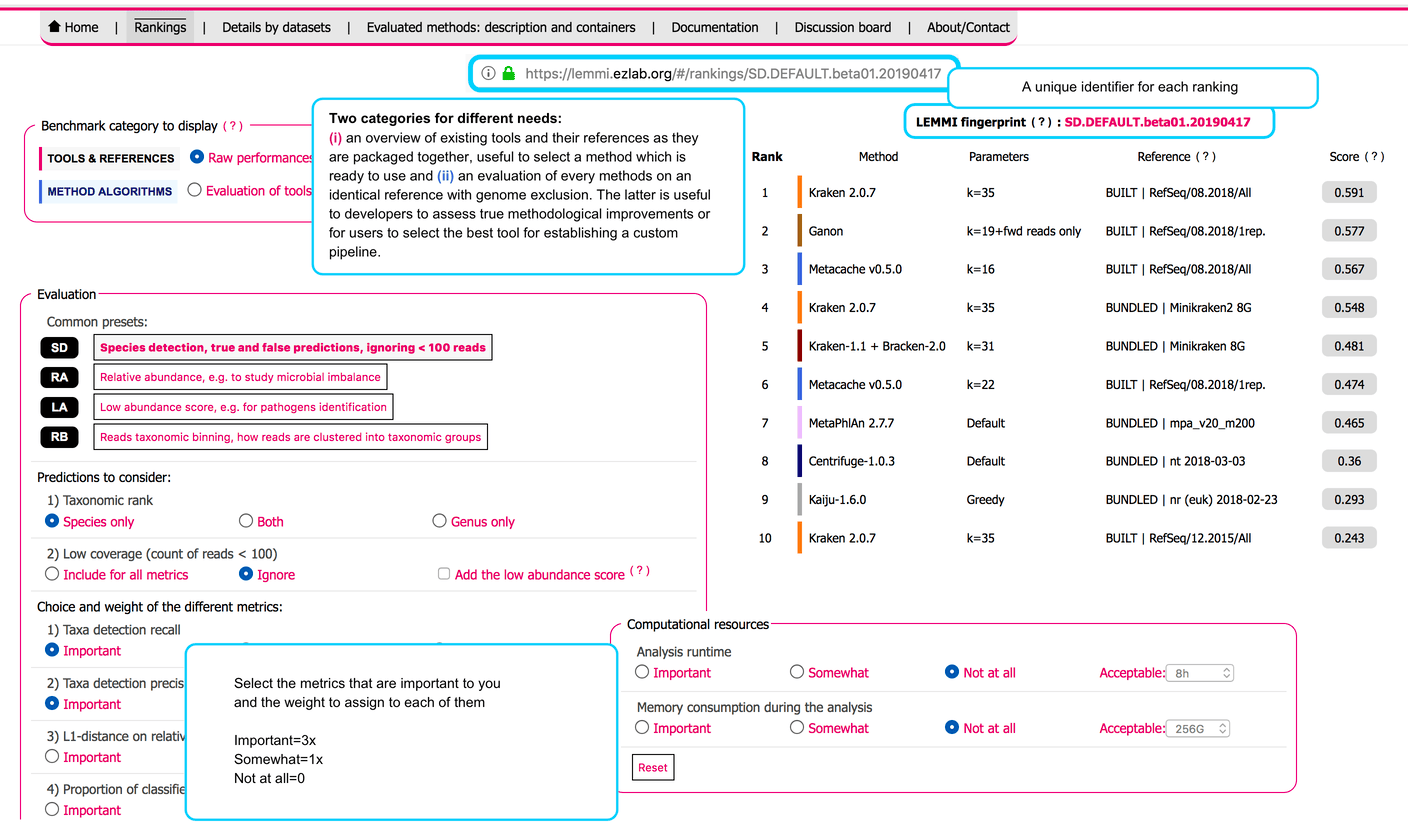
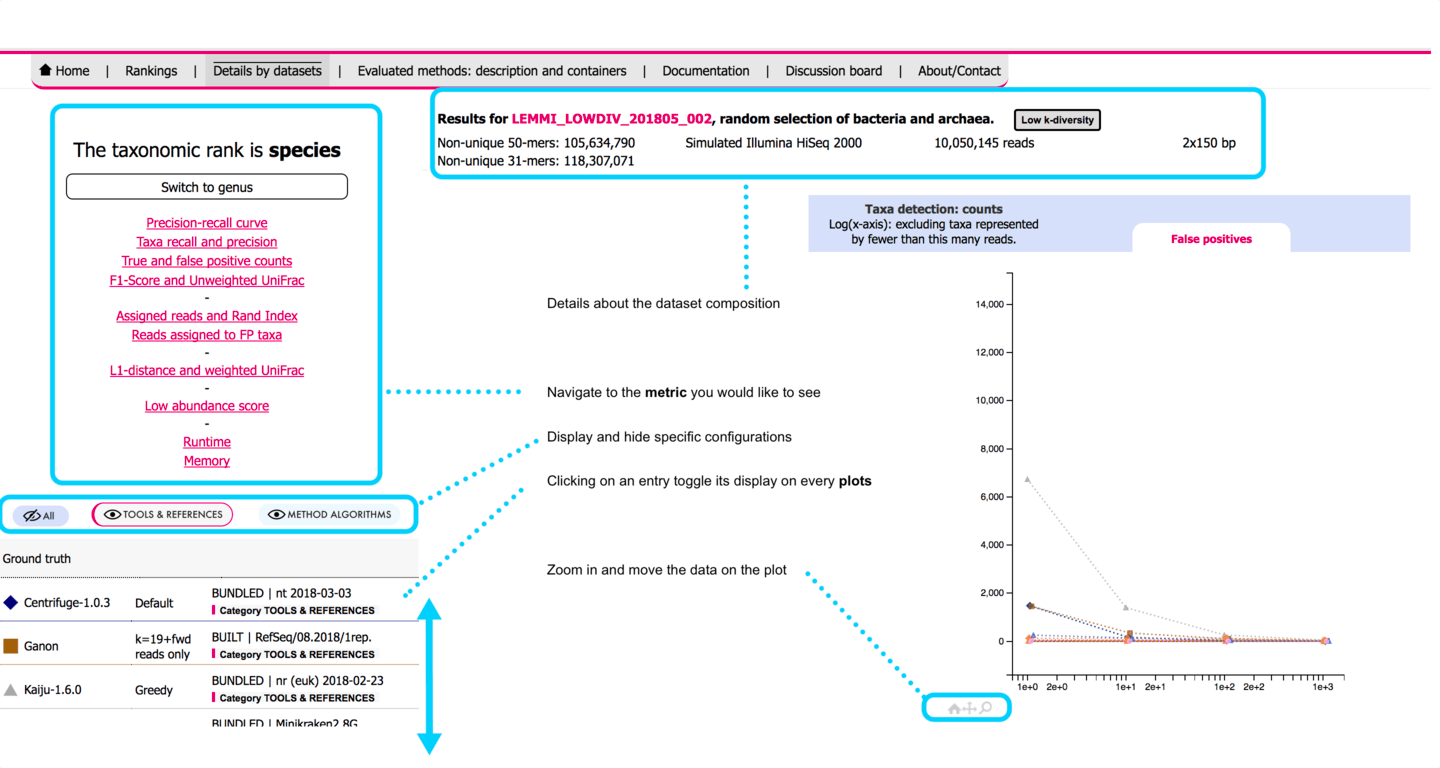
# User interface
# Workflow
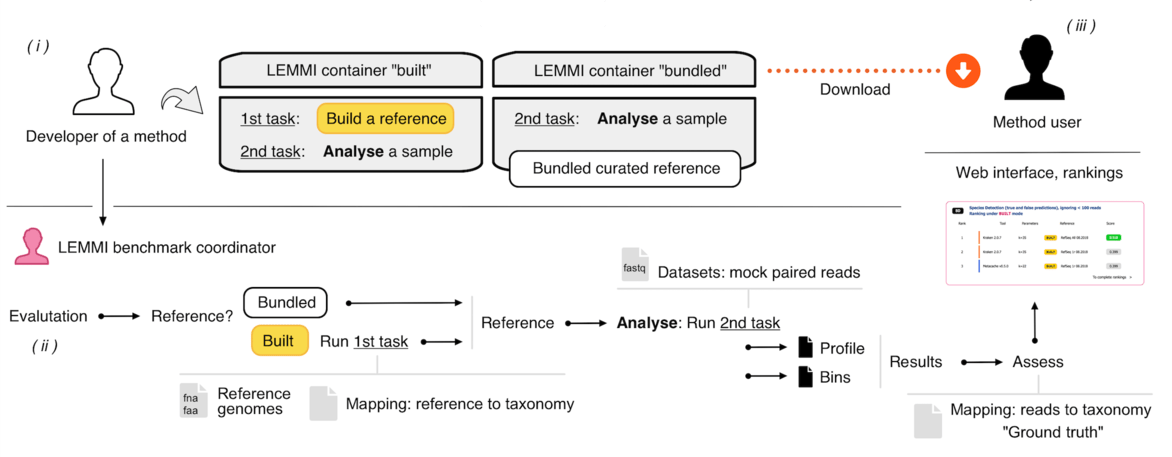
(i) Developers wrap their method in a container and test it on their own environment to ensure it works with the demo data. (ii) They submit the method for evaluation. The container goes through the benchmarking workflow to proceed to reference buildings and datasets analyses. (iii) After the results are evaluated against the ground truth, the method appears on the dynamic ranking on the LEMMI website.
# Two kinds of reference
BUILT identifies runs that are under the control of the LEMMI platform for the reference creation. The "build_ref" task of the container is called first, using genomic content provided by the LEMMI platform (i.e. RefSeq nucleotide assemblies or corresponding proteins). Moreover, the genome exclusion (BUILT+GE) approach allows the files used to generate the mock reads to be excluded from the files provided as reference for the analysis of the corresponding dataset. This requires multiple reference building, makes the challenge more difficult, and decreases the score of most methods. However, it represents the most reliable assessment of the true performances of a method.
BUNDLED identifies runs that used a reference provided by the creator of the container, which the LEMMI platform has no control on. All taxids reported by these references but unknown to the LEMMI platform are ignored. No metrics are available for the reference creation (i.e. memory and runtime).
# Two categories of benchmark
TOOLS & REFERENCES mixes BUILT and BUNDLED runs and is an overview of existing tools and their references as they are packaged together, useful to select a method which is ready to use or to investigate reference-specific questions. The heterogeneity of the references increases the variability of the results.
METHOD ALGORITHMS is an evaluation of every methods on an identical reference using genome exclusion (BUILT+GE). Useful to developers to assess true methodological improvements or for users to select the best tool for establishing a custom pipeline. Cannot include tools that do not provide scripts to build a reference.
# Results assessment
All outputs are post-processed to properly represent the evaluated taxonomic level. Binning results are then assessed with AMBER (opens new window) and profiles are assessed with OPAL (opens new window). Common metrics are extracted for all configurations, datasets, and for multiple low abundance filtering thresholds. Additional metrics are computed internally, for instance the low abundance score, the proportion of reads assigned to false positive taxa, and the memory and runtime values.
# Scoring scheme
All metrics that are not already values between 0.0 and 1.0, with 1.0 being the best score, are transformed. The L1 distance is divided by its maximum value of 2.0 and subtracted from 1.0, the weighted UniFrac score is divided by its maximum value of 16.0 and subtracted from 1.0. The unweighted UniFrac score is divided by an arbitrary value of 25,000 and subtracted from 1.0. The memory and runtime are divided by 2x the maximum value (as defined by the user through the interface) and subtracted from 1.0, to obtain a range between 0.5 and 1.0. This approach allows to segregate methods that remain below the limit from those that exceed it and get the value 0.0. Any transformed metric below 0.0 or above 1.0 is constrained back to the respective value. The final score displayed in the ranking is the harmonic mean of all metrics, taken into account zero, one, or three times depending on the weight assigned to the metric by the user.
# Low abundance score
The low abundance score is a custom metric calculated separately to evaluate the ability of the tool to correctly identify organisms present at very low coverage, but penalizing methods likely to recover them by recurrent report of the same taxids owed to very poor precision. To achieve this, as precision of low abundance organisms cannot be defined for a dataset (all false positives have a true abundance of zero and cannot be categorized as low abundance), the metric is computed by pairing two datasets to judge if a prediction can be trusted. The datasets (D1 and D2) include sets of taxa T1 and T2 that contain a subset of low abundance taxa (T1_low and T2_low, < 100 reads coverage, T1_low ≠ T2_low). Each taxon belonging to T1_low identified in D1 increases the low abundance score of the tool for D1 (recall) only when it is not identified in D2 if absent from T2. Otherwise, a correct prediction of the taxon in D1 does not improve the score (as proxy for low abundance precision). The score (0.0 - 1.0) is processed from both sides (D1, D2), to obtain an independent score for each dataset. This metric is only defined for all LEMMI_RefSeq datasets (low abundance species: n=10, n=8, for LEMMI_RefSeq_001 and LEMMI_RefSeq_002, respectively). A visual explanation exists in the LEMMI preprint (opens new window).
# Version control
The idea behind LEMMI is that users can obtain the tool as it appears on the ranking they are seeing. The fingerprint identifies a ranking that can be reloaded on the website at any time. Futhermore, the fingerprint also acts as a git tag (opens new window) to access to the sources corresponding to the containers at this version of the rankings. We are also planning to provide an archive of each container (i.e. to use with docker load < container.tar) in a way that can be linked to a specific fingerprint.
# Tutorial: prepare a method for benchmarking
To benefit from the visibility of the LEMMI platform and contribute to the establishment of a representative view of methods available to date, developers or advanced bioinformatics users need to wrap (or “containerize”) their metagenomic tool in a 'container' or 'box'.
If you are already familiar with the concept of container as implemented by http://www.bioboxes.org (opens new window) and had a look on the LEMMI preprint, you should directly have a try on the Kaiju container (opens new window) that is ideal to help you start your own. See also the files and folders. The taxonomic levels that have to be supported are genus and species.
Preparing a container is not a trivial task. If you need assistance and to plan the submission and assessment of your containerized method, please open an issue (opens new window).
# Docker containers and Bioboxes
LEMMI containers are based on the open-source Docker (opens new window) framework and, in part, on Bioboxes (opens new window), although not fulfilling all validation steps yet. These allow softwares to be installed and executed under an isolated and controlled environment, the so-called software containers, without affecting the host system from which the container is deployed, and allowing software to be easily shared and run free of dependencies issues and incompatibilities.
This guide shows you how to wrap your tool in a box that is compatible with the LEMMI pipeline.
Additional documentation can be found at https://docs.docker.com (opens new window) and http://bioboxes.org/docs/build-your-image/ (opens new window).
# Software requirements
To create a LEMMI box, you need to install Docker (see [https://docs.docker.com/install]/(https://docs.docker.com/install/)). To test if Docker has been correctly installed on your system try docker -v in your terminal.
# Host-container exchange files and folders, provided inputs, expected outputs
During the course of an evaluation, the LEMMI pipeline loads the candidate container one or several times. It mounts several folders present on its file system to be accessed by the container in its own file system as sub folders of /bbx/, to provide the fasta needed to build a reference, the mock reads to conduct an analysis, and several metadata files. The pipeline expects results in a specific formats and locations in the container file system. If the container is used for an actual analysis outside the LEMMI platform, the same file hierarchy has to be used.
# Input mapping and fasta
This mapping describes all input fastas (nucleotide and/or protein) provided for building the reference in /bbx/input/training/
/bbx/input/training/mapping.tsv
/bbx/input/training/GCF_123.1.fna.gz
No reliable information should be expected in the fasta header of individual files.
One file per line.
TAXPATH and TAXPATHSN may contain NA as follows: 2|1224|NA|NA|543|561|562
# LEMMI input mapping => genome for _ACCESSION = GCF_123.1 will be /bbx/input/training/GCF_123.1.fna.gz or .faa.gz
@Version:1.0.0
@Ranks:superkingdom|phylum|class|order|family|genus|species
@TaxonomyID:ncbi-taxonomy_2018-09-03
@@TAXID RANK TAXPATH TAXPATHSN _ACCESSION _SOURCE
562 species 2|1224|1236|91347|543|561|562 Bacteria|Proteobacteria|Gammaproteobacteria|Enterobacterales|Enterobacteriaceae|Escherichia|coli GCF_123.1 bacteria
# input names.dmp, nodes.dmp, and merged.dmp
provided in /bbx/input/training/
As found on NCBI: ftp://ftp.ncbi.nlm.nih.gov/pub/taxonomy/taxdump.tar.gz
# input reads.1.fq.gz reads.2.fq.gz
provided in /bbx/input/testing/
Fastq file of paired end short reads to analyze.
An example of LEMMI dataset can be found here:
https:doi.org/10.5281/zenodo.2651062 (opens new window)
# output bins.rank.tsv
A file in the CAMI binning format, written in /bbx/output/bins.$rank.tsv is expected.
Entries at the evaluated rank only are meaningful. Other entries will be ignored (no need to filter, and LEMMI pushes reads up to their parent level if needed).
If your method cannot provided binning of reads, an empty file (0 byte).
#CAMI Format for Binning
@Version:0.9.0
@@SEQUENCEID TAXID
read1201 123
read1202 123
read1203 131564
read1204 562
read1205 562
# output profile.rank.tsv
A file in the CAMI profiling format, written in /bbx/output/profile.$rank.tsv is expected.
Only the evaluated rank is required, although there's not need to filter other ranks.
If your method cannot provided relative abundance profile, an empty file (0 byte).
#CAMI Submission for Taxonomic Profiling
@Version:0.9.1
@SampleID:SAMPLEID
@Ranks:superkingdom|phylum|class|order|family|genus|species|strain
@@TAXID RANK TAXPATH TAXPATHSN PERCENTAGE
2 superkingdom 2 Bacteria 98.81211
2157 superkingdom 2157 Archaea 1.18789
1239 phylum 2|1239 Bacteria|Firmicutes 59.75801
1224 phylum 2|1224 Bacteria|Proteobacteria 18.94674
28890 phylum 2157|28890 Archaea|Euryarchaeotes 1.18789
91061 class 2|1239|91061 Bacteria|Firmicutes|Bacilli 59.75801
28211 class 2|1224|28211 Bacteria|Proteobacteria|Alphaproteobacteria 18.94674
183925 class 2157|28890|183925 Archaea|Euryarchaeotes|Methanobacteria 1.18789
1385 order 2|1239|91061|1385 Bacteria|Firmicutes|Bacilli|Bacillales 59.75801
356 order 2|1224|28211|356 Bacteria|Proteobacteria|Alphaproteobacteria|Rhizobacteria 10.52311
204455 order 2|1224|28211|204455 Bacteria|Proteobacteria|Alphaproteobacteria|Rhodobacterales 8.42263
2158 order 2157|28890|183925|2158 Archaea|Euryarchaeotes|Methanobacteria|Methanobacteriales 1.18789
# /bbx/reference/
This is the folder where the container has to write and read the reference, in any format compatible with the method, as long as everything is in this folder. What is created during the "build_ref" step will be available there during the "analysis" step.
# /bbx/tmp/
This is where the container has to write any temporary file and folder. Cannot be reused between "build_ref" and "analysis".
# Building a LEMMI container
To prepare all the files that are needed to build the box you can clone the Kaiju container (opens new window) and edit the files according to your needs while maintaining its structure.
# 1) Create a Dockerfile
In order to create and build a new box, you have to define a Dockerfile that contains all the instruction for creating the container.
# Define a base image
We recommend to use Ubuntu/Debian as a base image. You can specify this in the first line of your Dockerfile with:
FROM ubuntu:18.04d
Note that you can use a different distribution, or a different version by specifying it after the colon(😃.
# Install common dependencies
Now you can specify the dependencies that are required to install and run your metagenomic tool. For example if python3 and git are required and you are using Ubuntu, you have to specify in the Docker file the commands to install them. In this case you can use the apt-get install command after the Docker command RUN, as follows:
RUN apt-get install python3 -y
RUN apt-get install git -y
And for example, to install python modules you can install and use the pip installer:
RUN apt-get install python3-pip -y
RUN pip3 install pyyaml
RUN pip3 install jsonschema
RUN pip3 install scipy
# Install your metagenomic tool and dependencies:
You first need to copy the binary or source code of the tool from your local/remote machine or from a download/repository, here some examples:
# Using git:
ENV KRAKEN2_DIR /kraken2
RUN git clone https://github.com/DerrickWood/kraken2.git $KRAKEN2_DIR
WORKDIR $KRAKEN2_DIR
RUN git checkout 3167304affcff626db886c42c0ad3011965697a6
RUN ./install_kraken2.sh $KRAKEN2_DIR
RUN apt-get install ncbi-blast+ -y
Note: if you git clone, remember to git checkout a specific commit or tag for the sake of reproducibility.
You can then install the tool by executing the appropriate commands.
You can define variables with the ENV command, e.g.:
ENV KRAKEN2_DIR /kraken2
and add paths to the environment variable PATH with:
ENV PATH ${PATH}:$KRAKEN2_DIR:$KRAKEN2_DIR/bin
# Add additional files (entry, Taskfile, your scripts etc.)
You have to add all the files that will be used inside the box using the command ADD + filename + location (pay attention to the root of the box).
The following files are mandatory:
ADD entry /usr/local/bin/
ADD Taskfile /
entry should be reused untouched from the demo box (kaiju), and you will have to edit the Taskfile file (more on this below), but always add it at the root ( / ).
You then have to specify additional files such as scripts that are needed to process the input/output files, e.g.:
ADD scripts/prepare_inputs.py /
ADD scripts/prepare_inputs.sh /
ADD scripts/prepare_outputs.py /
ADD scripts/prepare_profiles.sh /
ADD scripts/prepare_profiles.py /
Remember to change permission to executable files (chmod +x bash_file_name) if you run them directly, e.g. ./script.sh.
Finally specify the starting point of when you call docker run analysis or docker run build_ref (more on this below).
The value from the demo box (kaiju) should be kept.
WORKDIR /bbx/tmp
ENTRYPOINT ["entry"]
# 2) Create/edit the Taskfile, overview
Once you have defined the instruction in the Dockerfile, you will have to edit the Taskfile to specify all the commands that are needed to run your metagenomic tool, and that will be executed when the container/box is launched.
The commands have to be defined under two tasks: "build_ref" and "analysis".
In the "build_ref" task you specify the commands required to build a reference that will be used when running the "analysis" task. To build the reference, the LEMMI platform provides fasta files (nucleotides or proteins) as input in /bbx/input/training/.
NOTE: if your metagenomic tool works only with a predefined reference you can skip this task and directly provide the external reference. On the LEMMI web platform this kind of benchmarking run will be tagged with the keyword “BUNDLED”, for provided reference.
The second task, "analysis" is where you specify the commands to actually run the metagenomic analysis, i.e. load and process the fastq reads to produce the output profile and binning file.
# Example
To build the reference starting from the list in /bbx/input/training/mapping.tsv:
build_ref:set -o verbose && mini=$(echo $parameter | cut -f2 -d",") && cd /kraken2 && alias python=python3 && mkdir -p /bbx/tmp/input/ && mkdir -p /bbx/reference/$reference/library/ && mkdir -p /bbx/reference/$reference/taxonomy/ && /prepare_inputs.sh && cp /bbx/input/training/*.dmp /bbx/reference/$reference/taxonomy/ && sleep 1 && /edit_fasta.sh && sleep 1 && /add_to_library.sh && kraken2-build --kmer-len $(echo $parameter | cut -f1 -d",") --minimizer-len=$(echo $parameter | cut -f2 -d",") --minimizer-spaces $((mini/4)) $(echo $parameter | cut -f3 -d",") --threads $thread --build --db /bbx/reference/$reference/
To run the analysis on the fastq files, reads.1.fastq.gz and its pair reads.2.fastq.gz, found in /bbx/input/testing:
analysis: set -o verbose && cd /kraken2 && alias python=python3 && /kraken2/kraken2 --fastq-input --threads $thread --paired --db /bbx/reference/$reference/ /bbx/input/testing/reads.1.fq.gz /bbx/input/testing/reads.2.fq.gz --output /bbx/tmp/tmp.out --report /bbx/tmp/tmp.out.report && python3 /prepare_results.py && mv /bbx/output/bins.tsv /bbx/output/bins.$taxlevel.tsv && mv /bbx/output/profile.tsv /bbx/output/profile.$taxlevel.tsv
# Some notes:
Always start the line with the name of the task followed by a colon (in this case build_ref:). Start the commands with “set -o verbose”, keeping all of your command on a single line, separating consecutive commands with “ && ”.
Avoid commands that have a limited argument size (e.g. cp, ls, etc.) for manipulating the input fasta files. Instead use find . -exec {};\, but put it in a separate bash file as escaping here may cause problems. For examples see the prepare_inputs.sh in the demo box (kaiju).
There are some parameters that must not be hard-coded in the commands specified in the Taskfile or either of the scripts called by the Taskfile. These are:
- the
$taxlevelvariable that defines the taxonomic rank on which the analysis focuses (species, genus, etc.) - the
$threadvariable which specify the number of cores to be used to run the analysis (max available 32). If your tool uses an amount of memory proportional to the number of core you may divide this value. 245GB of RAM is available.
You may want to pass additional parameters (e.g. k-mer size) as variables (not hard coding them in the commands), in order to be able to rerun a box with different parameters without rebuilding a new box for each combination of parameters.
The values of these parameters will be given when running the box in the $parameters variable. You can pass through this variable any number of parameters as a comma-delimited list. In the commands of the "build_ref" and "analysis" tasks or the scripts called by the two tasks, the parameter(s) of interest can be access by splitting the variable $parameters with a command like:
$(echo $parameter | cut -f1 d",") # this will return the first parameter in the comma-delimited list of parameters. E.g. parameters=23,greedy,foo,bar => 23
All inputs (*.fastq, *.fa, *.fna) are read-only, so they need to be gunziped as copy in /bbx/tmp/ if not read directly.
Apart from this, since there might be a huge amount of file to deal with as references, when possible try to edit files in place and not generate duplicates. Remove unnecessary intermediate files to save our server disk space.
Anything that is printed on stdout/sterr is reported in the final logs returned to the user.
# 3) Build the box
Once the Dockerfile, Taskfile and the required additional scripts have been prepared you can build the box. If you followed the example here your working directory (the folder from where you want to build the container) should look like this:
drwxr-xr-x scripts
-rw-r--r-- Dockerfile
drwxr-xr-x bbx
-rw-r--r-- Taskfile
-rw-r--r-- README.md
-rwxr-xr-x entry
You can then build the box by running the following docker command in you working directory (that will correspond to the root of your box):
docker build -t toolname --rm=true -–force-rm=true .
Avoid using “_” in toolname.
You should test the box on your machine before exporting it. A small test sample is packed with the kaiju container (opens new window) in the bbx folder that should be copy in your container directory, i.e. current working directory $(pwd).
As the LEMMI pipeline does not run containers with a root account, everything has to be accessible to an unprivileged user. To test, we suggest your current user uid, that can be obtained by the command uid. Here uid=1005.
If the name of the box is kraken2, and you want to run it at the species level and with a kmer size of 31, you can run a test to build the reference with:
docker run -e thread=4 -e taxlevel=species -e reference=myref -e parameter=31 \
-v $(pwd)/bbx/input/:/bbx/input:ro -v $(pwd)/bbx/output/:/bbx/output:rw \
-v $(pwd)/bbx/tmp/:/bbx/tmp:rw -v $(pwd)/bbx/metadata/:/bbx/metadata:rw \
-v $(pwd)/bbx/reference/:/bbx/reference \
-u 1005 kraken2 build_ref
You can check the log file in $(pwd)/bbx/metadata/log.txt.
And run the analysis with:
docker run -e thread=4 -e taxlevel=species -e reference=myref \
-v $(pwd)/bbx/input/:/bbx/input:ro -v $(pwd)/bbx/output/:/bbx/output:rw \
-v $(pwd)/bbx/tmp/:/bbx/tmp:rw -v $(pwd)/bbx/metadata/:/bbx/metadata:rw \
-v $(pwd)/bbx/reference/:/bbx/reference:ro \
-u 1005 kraken2 analysis
You can check the log file in $(pwd)/bbx/metadata/log.txt and the outputs in $(pwd)/bbx/output/. You should see profile.species.tsv and bins.species.tsv.
Once your LEMMI container is ready, package it as follows:
docker save toolname > toolname.tar
gzip toolname.tar
If you need a custom reference folder, package it as follows in a separate file containing a unique folder named after your reference (i.e. ./myreference_folder): toolname.reference.tar.gz
You are now ready to submit the box.
# 4) Submit the box
See the FAQ below.
# FAQ
# How can I submit my tool?
First, open an issue (opens new window) to help planning and tracking your submission, and follow our tutorial to wrap your method in a container. Then, you can either fork the lemmi project (opens new window) to add the source of your container in the methods_submission branch and submit a merge request, so we can build an run it, or share on https://hub.docker.com/ (opens new window) a built container that we can pull and run.
If additional information are needed, or a more confidential exchange of container is required, contact us directly by email (ez at ezlab.org)
# You included in the list the tool I develop and I think it could be better used. Can I re-submit it?
We encourage it, as we think that tool developers may be able to make technical choices that will show the best potential their method can offer.
# I develop a commercial product. Is it possible to benchmark it without releasing the source or the archive?
Yes. We aim at providing an overview of all existing solutions available to users. Therefore, if a solution that appears in the ranking cannot be distributed by LEMMI, it will be just indicated that the software is not freely available.
# My tool cannot build a reference on demand, can it still be included in the ranking?
Yes, in this case we require you to provide a reference on your own and it will be ranked only under the TOOL & REFERENCES category.
# What are the input and output files supported by the containers?
Our platform uses the CAMI binning and profiling formats. See the detail in the user guide and the kaiju container (opens new window)
# Which version of the taxonomy is used?
A specific version of the NCBI taxonomy is used at each major release. The version used in beta01 was downloaded the 03/09/2018.
# Is the NCBI taxonomy the only one supported?
It is for beta01, but LEMMI envisions to open the benchmark to alternative taxonomies in a future release, potentially assessing the impact of the taxonomic system itself on the results. We strongly encourage tool developers to structure their software in a way that allows switching between taxonomies.
# What does relative abundance in LEMMI means?
Tools report abundance of reads or abundance of organisms (proxied by number of markers, genomes) depending on their methodology, which is not directly comparable. To avoid fragmenting the methods into two categories, we think results should be harmonized. LEMMI expects the number of organisms to be reported, defined by normalizing the number of reads to genome average sizes. Since reads abundance can be converted if the genome size is recorded during the reference creation process, this approach enables the evaluation of read classifiers and marker/assemblies based profilers together. LEMMI does not convert results automatically as its goal is to reflect the native performances of all presented tools on that question. Instead, LEMMI promotes the idea of adding a normalization layer on the top of read profilers in future submissions, which should improve performances on abundance estimation in this class of tools. It also favors progress towards standardized outputs for tools serving similar purposes or often considered as such by users. It is worth noting that in LEMMI beta01, focused on bacteria and archaea, large differences of taxa recall will affect the abundance prediction more negatively than an absence of normalization from reads to organisms.
# Can I obtain the source code of the containers presented on the LEMMI website?
Yes, see https://gitlab.com/ezlab/lemmi/tree/master/containers (opens new window) and version control.
# Can I run the LEMMI pipeline on my own server?
Not in the current version, but a standalone version that enables private benchmarking while developing is envisioned in the future